
Digitalisierung
Modellierung biomolekularer Systeme bietet die Möglichkeit, Verhalten von Proteinen auf Molekül- und Prozessebene besser zu verstehen. Wir beschäftigen uns mit zwei unterschiedlichen Ansätzen. Mit der Moleküldynamiksimulation (MD Simulation) können einzelne Proteine in ihrer atomaren Umgebung simuliert und deren Verhalten während diverser Aufreinigungsschritte vorhergesagt werden. Mit der mechanistischen Modellierung chromatographischer Systeme wird der Aufreinigungsprozess makroskopisch betrachtet. Transport, Bindung und Diffusion werden mit Differentialgleichungen beschrieben. Hiermit wird eine optimale Prozessauslegung ermöglicht.
Process analytical technology (PAT)
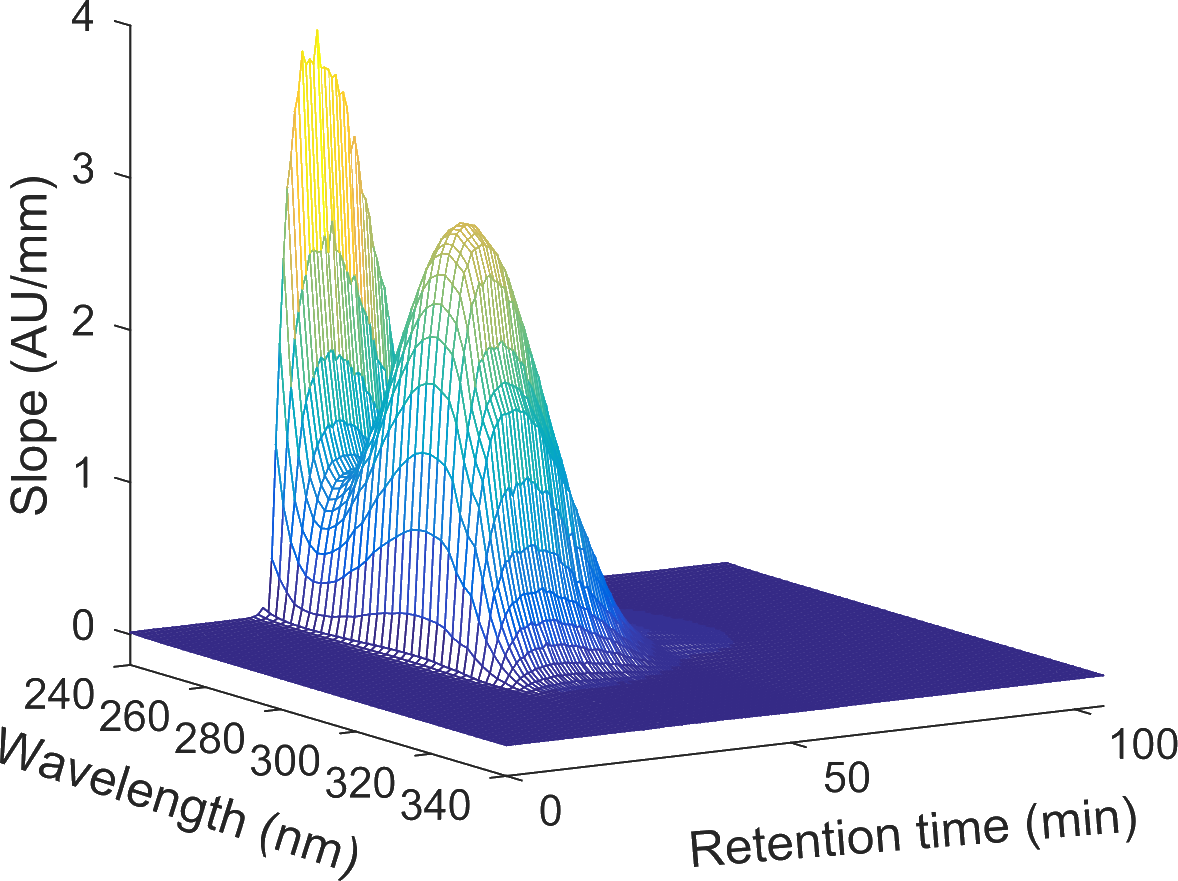
In der Produktion von therapeutischen Proteinen ist eine effiziente Prozessüberwachung für einen ökonomischen Prozess entscheidend. Gegenwärtig beschränkt sich die online Prozessüberwachung in der pharmazeutischen Industrie auf univariante Prozessparameter wie pH, Leitfähigkeit oder Absorption des Effluents. Erst in offline Analysen werden zentrale Prozessattribute (z.B. Zielproteingehalt, Konzentration an koeluierenden Kontaminaten, etc.) bestimmt. Diese Vorgehensweise ist nicht nur zeitaufwendig, sondern kann auch dazu führen, dass Inkonsistenzen in der Produktion erst spät erkannt werden und unter Umständen gesamte Batches verworfen werden müssen. In den letzten Jahren hat das Interesse der pharmazeutischen Industrie an der Erfassung zentraler Prozessparameter in Echtzeit zugenommen. Besonders die QbD und PAT Initiative der amerikanischen Gesundheitsbehörde FDA haben starke Anreize zur Entwicklung solcher Technologien gesetzt.
Die Gruppe Molekulare Aufarbeitung von Bioprodukten befasst sich mit dem Einsatz chemometrischer Methoden für die Überwachung von chromatographischen Prozessen zur Proteinaufreinigung. Wir forschen auf folgenden Gebieten:
- Prozessüberwachung mittels Spektroskopie in Kombination mit chemometrischen Methoden
- Echtzeitprozesskontrolle basierend auf den gewonnen Informationen
- Rückwirkende Fehleranalyse
Literatur (zum Ausklappen klicken)
Data science and data visualization
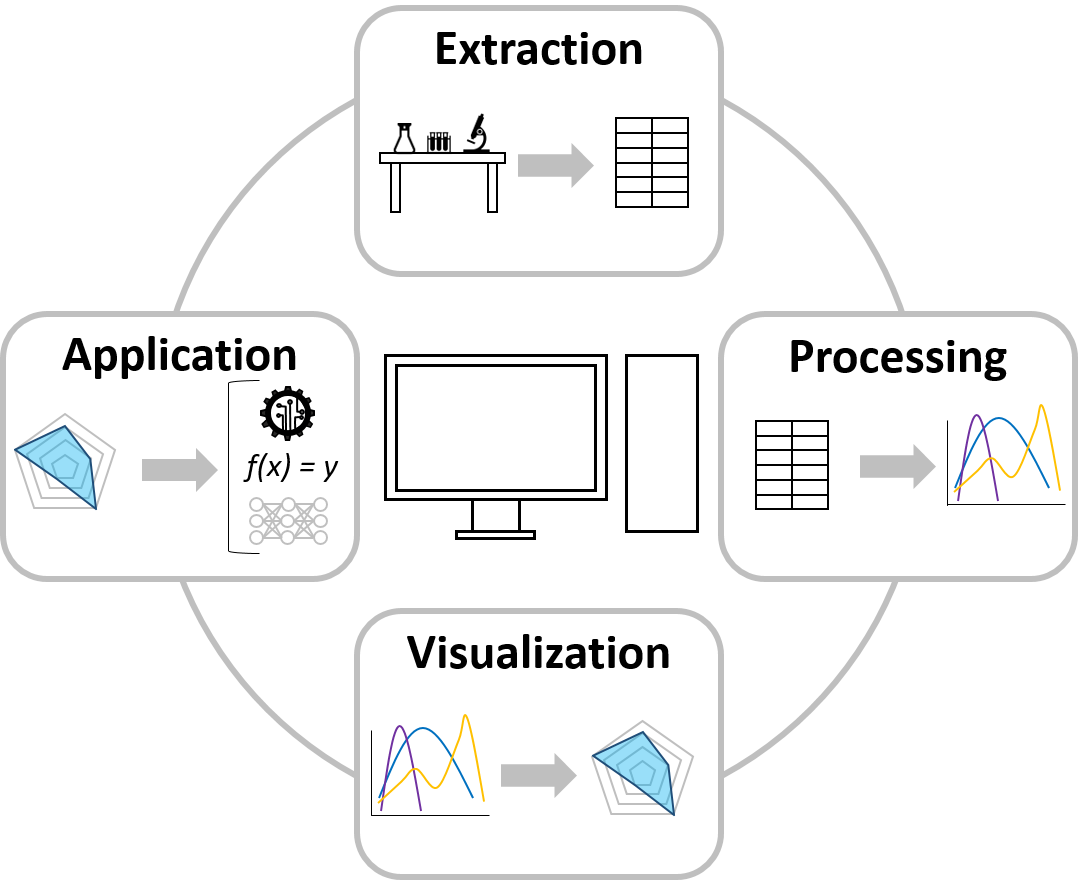
Der Bereich Datenwissenschaft und Visualisierung arbeitet an fortgeschrittenen Ansätzen zur Etablierung und Verbindung von Datenverarbeitungsschritten wie Extraktion, Verarbeitung, Visualisierung und Anwendung. Im MAB streben wir mit Hilfe dieser datenwissenschaftlichen Strategien ein breiteres Verständnis der Bioverarbeitung an. Dazu gehören automatisierte Datenextraktionsprotokolle für hauseigene Laborgeräte und die anschließende Datenbereinigung und -verarbeitung, die für die weitere Verwendung erforderlich sind. Es werden Algorithmen etabliert, um die verarbeiteten Daten mittels (multidimensionaler) Datenvisualisierung zu präsentieren, um Datenmuster in großen und sonst unübersichtlichen Datensätzen darzustellen. Die Datenanwendung wird durch die Entwicklung von Algorithmen für maschinelles Lernen und tiefes Lernen für Klassifizierungs- und Regressionsprobleme realisiert, die zur Erstellung von prädiktiven und präskriptiven Modellen verwendet werden können.
Literatur (zum Ausklappen klicken)
Mechanistische Chromatographiemodellierung
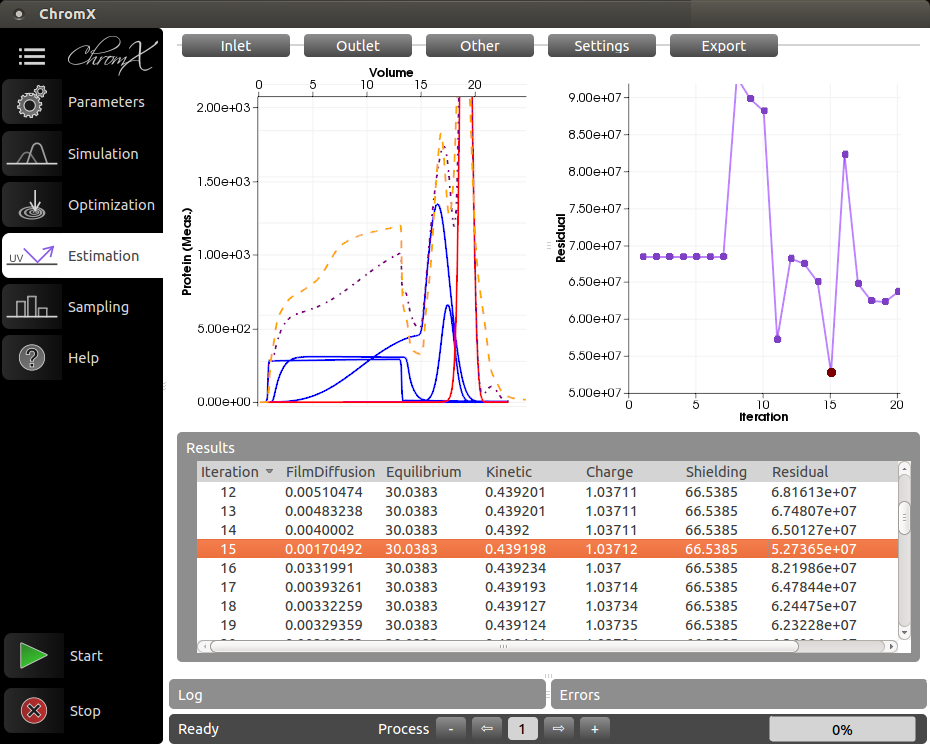
Präparative Chromatographie ist eine der wichtigsten Technologien für die Aufreinigung von Biologics, wie z.B. therapeutischen Proteinen. Die modellbasierte Chromatographie-Prozessentwicklung ist die logische Reaktion auf den Quality-by-Design Ansatz, vorgeschlagen von amtlichen Behörden wie FDA, EMA usw. Ein mechanistisches Modell, bestehend aus Differentialgleichungen unterschiedlicher Komplexität, beschreibt die Stoffübertragung und Adsorptions-/Desorptionsreaktionen innerhalb der Chromatographiesäule. In den letzten Jahren wurde die intern entwickelte Simulationssoftware ChromX für die mechanistische Modellierung erfolgreich eingesetzt, um VLPs, monoklonale Antikörper und andere Proteine aufzureinigen bzw. die Aufreinigung zu optimieren. Durch die modellbasierte Prozessentwicklung können bis zu 95% der Laborversuche für Prozessentwicklung, Robustheitsstudien und Worst-Case-Analysen durch computergestützte Simulationen ersetzt werden. Die dabei gewonnenen mechanistischen Prozessverständnisse sind vom großen akademischen Interesse.
ChromX
ChromX ist eine Simulationssoftware für Proteinchromatographie. Sie wurde ursprünglich am MAB entwickelt, wird inzwischen aber durch eine unabhängige Ausgründung weiterentwickelt. Mehr Informationen gibt es unter GoSilico GmbH.
Literatur (zum Ausklappen klicken)
3D-Strukturbestimmung
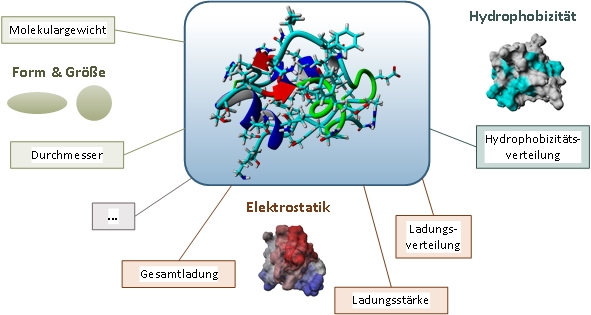
Durch die Entwicklung leistungsstarker Computer mit Parallelarchitektur ist es möglich geworden, die Dynamik molekularer Systeme zu simulieren. Die Moleküldynamiksimulation (MD) von Proteinen bietet vielfältige Möglichkeiten: von der Reduktion des experimentellen Aufwands, über den Zugang zu experimentell schwer zugänglichen Größen bis hin zu einem tieferen Verständnis für das Verhalten komplexer Systeme. Der am Institut entwickelte, automatisierte Arbeitsablauf von MD-Simulationen umfasst einige Schritte, bei denen das Molekül in Ziel-Prozessbedingungen (z.B. pH und Ionenstärke) gebracht und datenabhängig simuliert wird. Diese 3D-Strukturen sind auch Ausgangspunkt für die Anwendung von quantitativer Struktur-Wirkungsbeziehung (QSAR). Das Ziel von QSAR ist die Vorhersage von chemischen sowie biologischen Eigenschaften und Aktivitäten noch nicht synthetisierter Substanzen. Dieser Theorie liegt die Annahme zugrunde, dass Eigenschaften und Aktivitäten komplett durch die molekulare Struktur eines Proteins bestimmt werden. Diese wird durch verschiedene Deskriptoren, wie beispielsweise Form, Größe, Elektrostatik und Hydrophobizität beschrieben. Diese Daten bilden den Ausgangspunkt der multivariaten Datenanalyse, welche molekulare Eigenschaften bekannter Strukturen auf Basis geeigneter Deskriptoren und deren experimentelles Verhalten in Zusammenhang bringt und dabei prädiktive Modelle erstellt, mit denen die Eigenschaften und Aktivitäten für neue Moleküle vorhergesagt werden können.