
Digitalization
Modelling of biomolecular systems offers the possibility to better understand the behaviour of proteins at the molecular and process level. We are dealing with two different approaches. Molecular dynamics simulation (MD simulation) allows us to simulate individual proteins in their atomic environment and to predict their behavior during various purification steps. With the mechanistic modeling of chromatographic systems, the purification process is viewed macroscopically. Transport, binding and diffusion are described using differential equations. This enables an optimal process design.
Process analytical technology (PAT)
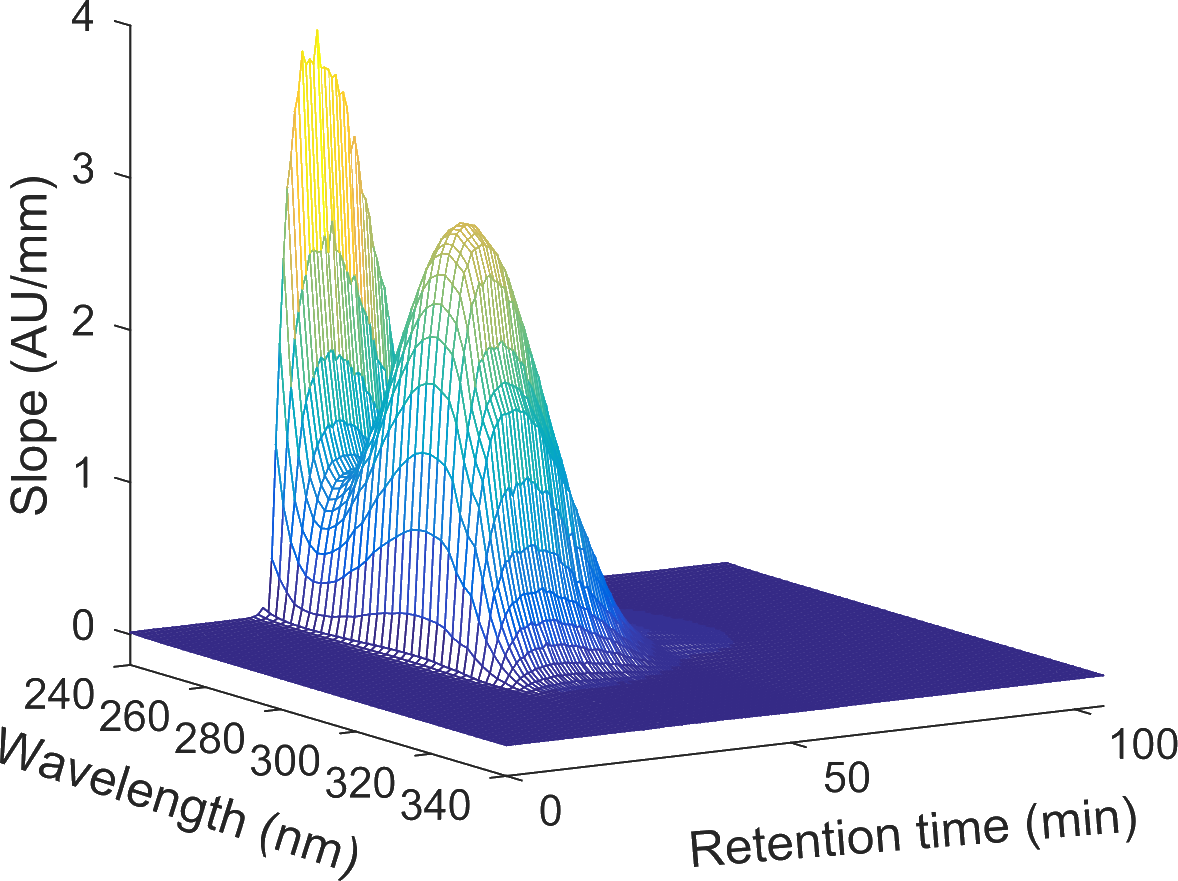
In the production of biopharmaceuticals, high and consistent product quality is key. However, in current downstream processes, real-time process monitoring and control play a minor roll in assuring the product quality. Instead, industry widely relies on offline analytics to ensure product quality (e.g. product content, concentration of co-eluting contaminants, host cell proteins) and to define important process parameters. This approach is however time-demanding and may introduce unwanted variability into the process.
In 2004, US Food and Drug Administration (FDA) introduced the Process Analytical Technology (PAT) initiative, which aims to promote (near) realtime process monitoring and control. The research group MAB works on chemometric methods for realtime monitoring. A special focus is set on:
- Process monitoring by spectroscopy in conjunction with chemometrics
- Realtime process control based on the obtained information
- Root-cause investigations
Literature
Data science and data visualization
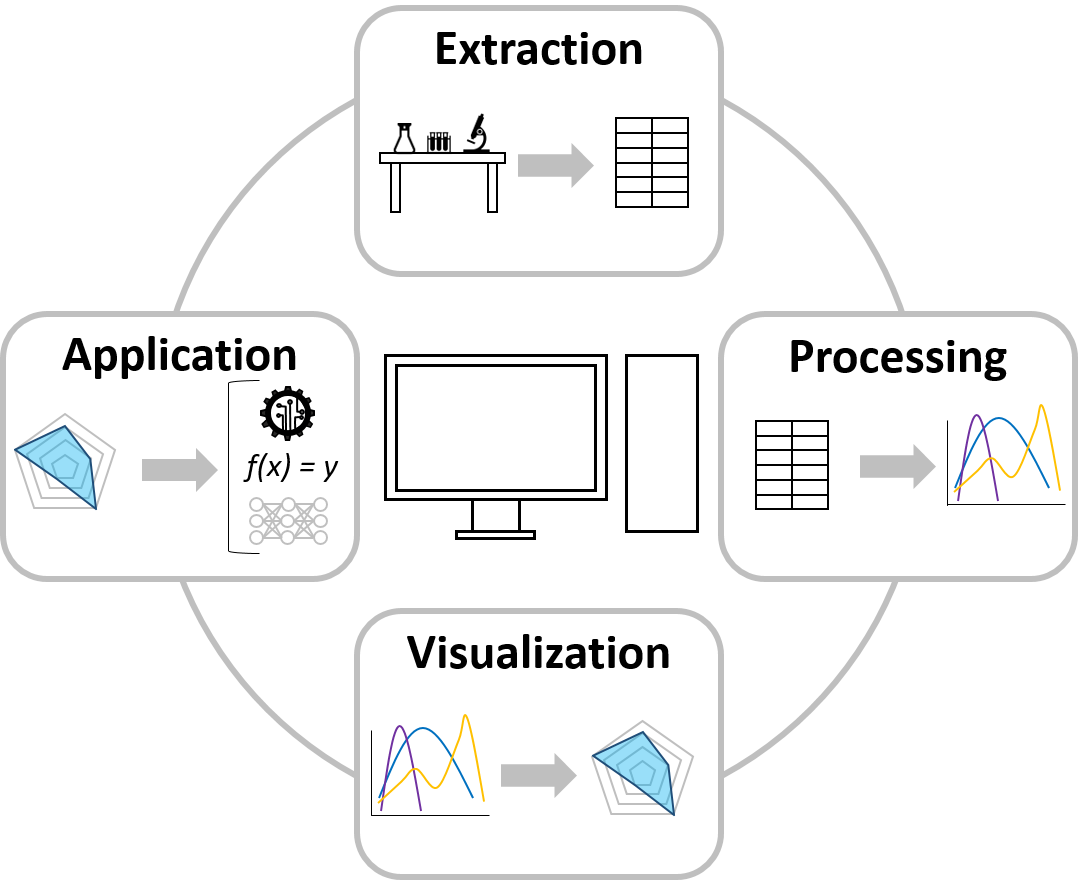
The field of data science and visualization works on advanced approaches to establish and connect data handling steps such as extraction, processing, visualization, and application. At MAB we strive to generate a broader understanding of bioprocessing by means of these data science strategies. This includes automated data extraction protocols for in-house labware and subsequent data cleaning and processing required for further usage. Algorithms are established to present the processed data by means of (multidimensional) data visualization to display data patterns in large and otherwise incomprehensive data sets. Data application is realized by developing machine learning and deep learning algorithms for classification and regression problems, which can be used to establish predictive and prescriptive models.
Literature
Mechanistic Modeling of Chromatography
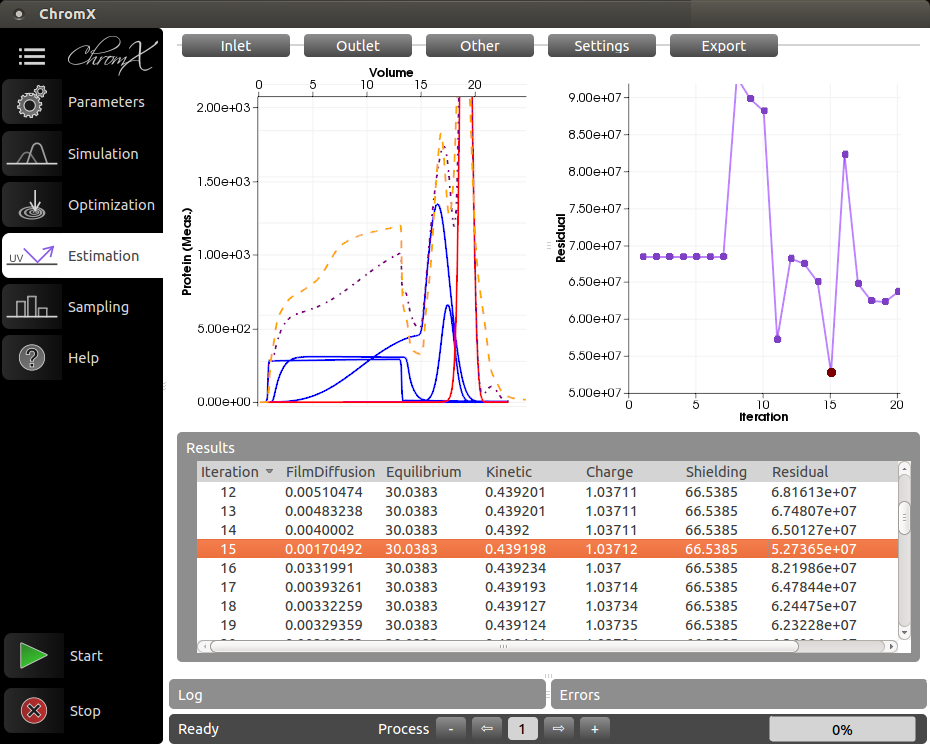
Preparative chromatography is one of the most important technologies for the purification of biologics, e.g. therapeutic proteins. Model-based chromatography process development is the response to Quality-by-Design (QbD) approach required by regulatory authorities such as FDA, EMA, etc. A mechanistic model consists of differential equations of varying complexities, which describe the mass transfer effects and the protein-ligand interaction within a chromatography column. In the last years, the in-house developed software ChromX could be successfully used for model-based process development to purify virus-like particles, monoclonal antibodies and other proteins. The benefits of model-based process development are obvious: up to 95% lab experiments for process optimization, robustness studies, and worst-case analysis can be replaced by computer-aided simulations. From the academic point of view, the gain of mechanistic understanding of the process is a great advantage.
ChromX
ChromX is a simulation toolbox for liquid chromatography of proteins. The conceptual goal of ChromX is to be a flexible multi-purpose software package for research while maintaining a high level of user-friendliness. ChromX was originally developed at MAB and is now commercialized by the startup GoSilico GmbH.
Literature
3D-Structure determination
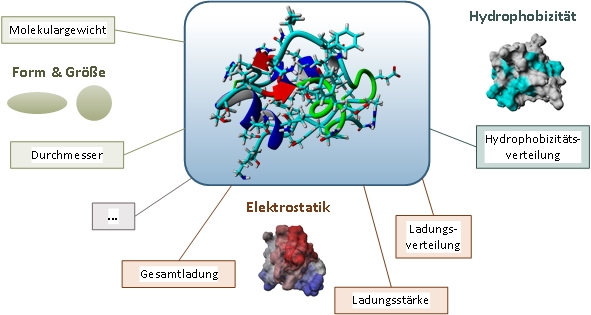
By developing powerful computers with parallel architecture it has become possible to simulate the dynamics of molecular systems. The Molecular dynamics simulation (MD) of proteins offers a wide range of possibilities: from the reduction of the experimental effort, to access to experimentally difficult to access sizes up to a deeper understanding of the behavior of complex systems. Researchers at the institute developed an automated workflow of MD simulations includes some steps in which the molecule is immobilized in target process conditions (e.g. pH and ionic strength) and simulated in a data-dependent manner. These 3D structures are also the starting point for the application of quantitative Structure-activity relationship (QSAR). The goal of QSAR is the prediction of chemical and biological properties and activities not yet of synthesized substances. This theory is based on the assumption that properties and activities completely through the molecular structure of a protein can be determined. This is def ined by various descriptors, such as shape, size, electrostatics and hydrophobicity. These data form the starting point for multivariate data analysis, which molecular properties of known structures based on suitable descriptors and their experimental behaviour and predictive models are created with which the properties and activities for new molecules can be predicted.